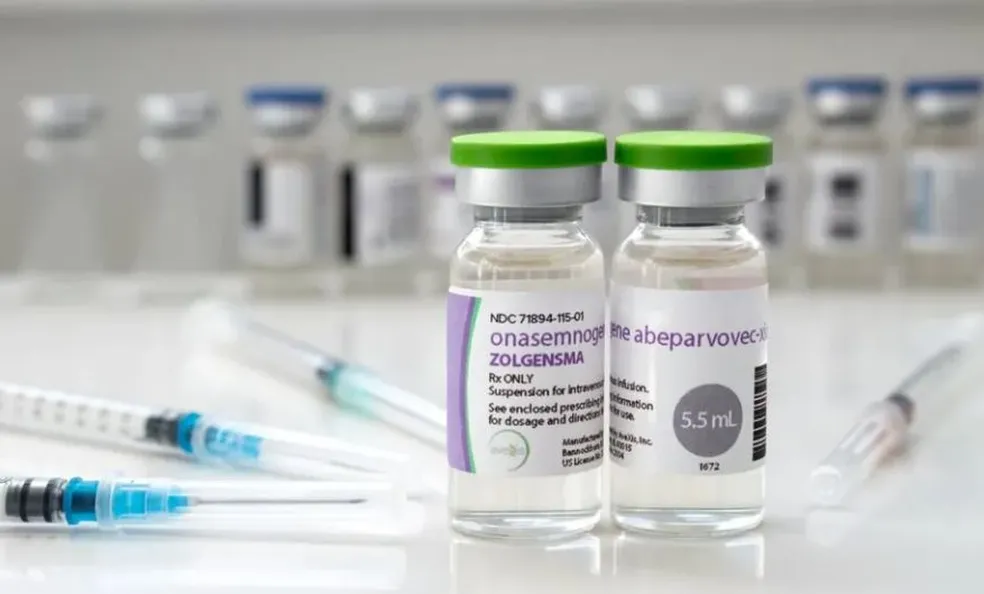

O Sistema Único de Saúde (SUS) já incorporou três medicamentos para o tratamento da atrofia muscular espinhal (AME). O primeiro, que passou a ser oferecido em 2021, é o Spiranza (Nusinersena), aprovado para os tipos 1 e 2 da doença (até 18 meses de vida). O medicamento de R$ 320 mil cada dose tem representado uma melhor qualidade de vida também para pacientes jovens, como a cantora Daniela Bublitz, de 29 anos – retratada aqui no Portal ViDA & Ação no Dia Internacional da Mulher (8 de março).
Depois vieram a solução oral Evrysdi (Risdiplam) – também para os tipos 1 e 2 – e a terapia gênica Zolgensma, exclusivamente para o tipo 1 (bebês de até 6 meses). Juntas, as três medicações significam uma esperança a mais para controle e até cura definitiva de uma doença rara e grave que causa sérios comprometimentos e pode levar a criança à morte até dois anos de vida.
Apesar de aprovados pelo Ministério da Saúde, ainda há muita dificuldade de acesso por causa da burocracia (entenda melhor abaixo). Enquanto isso, pacientes que têm pressa em usar o medicamento precisam entrar com ação na Justiça ou custear a compra, muitos com ajuda de campanhas de doação (‘vaquinhas’) que circulam na internet.
Segundo a fabricante Novartis, desde sua aprovação pela Agência Nacional de Vigilância Sanitária (Anvisa) em agosto de 2020 e disponibilização no mercado brasileiro, 107 pacientes foram tratados com Zolgensma no Brasil. O medicamento já foi considerado o mais caro do mundo – hoje é o segundo e custa em torno de R$ 6 milhões a dose única.
Tratamentos surtem mais efeitos em bebês até 6 meses e sem sintomas
Apesar de o Brasil já contar com três tipos de medicamentos aprovados para AME, especialistas explicam que o tratamento da doença rara, degenerativa e sem cura ainda surte mais efeitos em bebês de até seis meses de idade que são diagnosticados antes de surgirem os sintomas. Por isso, a luta das famílias para acelerar a cobertura da AME no Teste do Pezinho ampliado, visando ao diagnóstico precoce e, assim, garantir o acesso de todos os pacientes aos medicamentos disponíveis no SUS.
A AME é uma das 53 patologias raras que deverão ser incluídas na implantação progressiva do Teste do Pezinho ampliado no SUS em todo o país, mas ainda não se sabe quando – o prazo final é 2027. A Lei 14.154, de 26 de maio de 2021, estabeleceu a ampliação – de seis para 53 – do número das doenças que podem ser detectadas pela triagem neonatal, conhecida como Teste do Pezinho, que permite o diagnóstico precoce. A lei federal passou a vigorar em 27 de maio de 2022 e deu aos estados prazo de quatro anos para a incorporação de todas as enfermidades.
Embora a AME esteja relacionada na quinta e última etapa de implantação do Teste do Pezinho ampliado, não há ainda previsão de quando ela será efetivamente incorporada ao SUS. Um movimento tenta sensibilizar os estados para que se mobilizem e acelerem a implantação das novas doenças na triagem neonatal, o que poderá beneficiar diretamente pacientes de AME na fase inicial da doença.
Tratamentos promissores para recém-nascidos pré-sintomáticos
Para o Universo Coletivo AME, uma união de cinco instituições que atuam há mais de 20 anos em diferentes regiões do país e são lideradas por mães que vivenciam a AME no dia a dia, os tratamentos hoje disponíveis no SUS são promissores, caso administrados em recém-nascidos pré-sintomáticos. Daí a luta das famílias e, especificamente, do grupo, para a inclusão da enfermidade no diagnóstico feito pelo Teste do Pezinho.
De acordo com especialistas, se não diagnosticada logo nos primeiros dias de vida, a AME pode comprometer o funcionamento do sistema nervoso motor e dos músculos de forma acelerada. “Quanto antes se inicia o tratamento menos terapias e homecare a criança precisará”, destaca o Universo Coletivo AME. Em setembro de 2023, mostramos aqui casos em que famílias chegam a gastar R$ 15 mil por mês com o tratamento da AME.
A neurofisiologista e neurologista Marcela Câmara Machado, membro da Academia Brasileira de Neurologia (ABN), diz que todas as medicações por via terapia gênica – como Zolgensma – têm muito mais efeito se a criança ainda não apresenta sintomas. “Ou seja, a gente diagnostica logo que ela nasce, antes de manifestar os sintomas, para ter uma vida, senão normal, muito próxima do normal”, disse.
Segundo ela, “os tratamentos são para otimizar e dão melhor resultado se os diagnósticos são feitos precocemente. Mas ainda não tem cura, apesar dessa terapia genética que se propõe a melhorar a função do indivíduo, recompor a função que é perdida, do ponto de vista genético”.
A gente não sabe, inclusive, se essa terapia genética terá de ser repetida na pessoa na fase adulta, por exemplo. Não há ainda estudos para isso. O que se sabe, atualmente, é que quanto mais precoce a criança é tratada, ela tem um desfecho melhor”, destacou a médica.
Novos tratamentos evitam progressão da doença
Os avanços no entendimento molecular da condição de AME culminaram com o surgimento de terapias inovadoras, ora atuando no principal modificador do fenótipo, o gene SMN2, ora com a utilização de terapia gênica, introduzindo nas células do paciente um novo gene com a mesma função do SMN1, conseguindo, no momento atual, modificar a história natural da doença”, destaca a especialista.
Teste da Bochechinha: exame genético para AME só na rede privada
A neuropediatra explica que a AME – causada pela perda irreversível dos neurônios motores na medula – é uma doença grave e fatal com grande impacto econômico e social. Diante deste cenário, a testagem e o diagnóstico precoce são cruciais para salvar a vida das crianças. Por isso, é necessário implantar a triagem neonatal para a doença em todo o território nacional.
O objetivo é diagnosticar bebês com AME em uma fase anterior ao início das manifestações clínicas da doença para que, assim, mais vidas sejam salvas. A conscientização e engajamento da população sobre a urgência da inclusão da AME na triagem neonatal da rede pública é essencial para salvar a vida de muitas crianças e buscar alterar o curso natural da doença no País”, afirma.
Dra Vanessa explica que pacientes com características clinicas de AME devem ser imediatamente encaminhados para o exame genético. Porém, atualmente, só é possível fazer o exame para AME na rede privada de saúde, por meio do chamado Teste da Bochechinha.
O exame é feito com uma amostra de saliva, colhida com uma espécie de cotonete na parte de dentro da bochecha. O teste fornece o DNA e acusa alterações com grande probabilidade de provocar doenças. O exame custa em média R$ 2 mil em clínicas especializadas em testes genéticos.
Além de salvar vidas, economia para os cofres públicos
A engenheira Adriane Loper criou o Instituto Fernando, em homenagem ao filho que morreu aos 9 anos de idade, após passar toda a vida internado em uma UTI, em decorrência da doença. Uma das líderes do Universo Coletivo AME, ela diz que das 50 doenças listadas e que estão à frente da AME, muitas ainda não têm tratamento. Já a AME, segundo ela, não precisa de tecnologias que tenham que ser adquiridas.
“É uma questão de reagentes, mas não de tecnologias”, comentou. Ela citou, ainda, que em congresso realizado em 2023 nos Estados Unidos, foram apresentados casos de crianças que, em função da triagem neonatal, feita nos últimos anos, tomaram medicação com oito dias de vida “e estão andando, com todos os marcos motores normais”.
Ela defendeu que a AME seja incorporada logo no Teste do Pezinho, para que outras crianças, como seu filho, tenham direito à vida. Disse também que as crianças tratadas a partir do diagnóstico precoce representarão uma “economia gigante” para o poder público.
Nos Estados Unidos, conforme pesquisas apresentadas no congresso, os custos com tratamento precoce da AME se tornaram sete vezes menores. “Se a gente não consegue diagnosticar precocemente, não vai conseguir tratá-las com essa terapia”, ressaltou a médica.
Triagem neonatal é essencial para detectar doenças genéticas
Introduzido no Brasil na década de 70, tornou-se obrigatório com a instituição da portaria do Programa Nacional de Triagem Neonatal (PNTN) em 2001 pela Lei número 822, incorporado pelo Governo Federal. O teste tem o objetivo de detectar precocemente doenças genéticas, infecciosas e hematológicas e é fundamental para o início do tratamento específico e precoce.
Porém, existe uma doença muito grave, a Atrofia Muscular Espinhal (AME), que ainda não está contemplada no PNTN Para Aline Giuliani, presidente do Instituto Viva Iris, falar da importância da triagem neonatal e da incorporação da AME no exame do Sistema Único de Saúde é urgente, necessário, e de muita relevância.
Sabemos que ela ainda é a maior causa genética de óbito em crianças menores de dois anos de idade, então, a pergunta deveria ser: por que não incluir a AME? Não vemos respostas. É uma condição que atende a todos os critérios para inclusão no programa de triagem neonatal”, complementa Aline.
Através deste teste é possível proporcionar para muitas crianças e famílias uma vida com qualidade e proteção sobre sérias circunstâncias impostas por doenças altamente impactantes. A Atrofia Muscular Espinhal também pode ser detectada e tratada precocemente, o que mudaria completamente o paradigma das consequências desta doença”, declara.
Entenda a burocracia para incluir novos tratamentos da AME no SUS
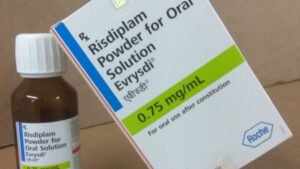
Enquanto Spinraza é dado por meio de injeções na lombar a cada quatro meses, Evrysdi (Risdiplam), produzido pela Roche, é uma solução oral e deve ser tomado todo dia. Ambos são de uso contínuo. Já Zolgensma é uma terapia gênica que deve ser administrada em dose única, o que explicaria seu altíssimo custo. Este medicamento chegou a ser considerado o mais caro do mundo.
Em 2021, Zolgensma custava US$ 2,1 milhões (ou cerca de R$ 10,5 milhões). Mas perdeu o ‘posto’ em 2022 para o Hemgenix, indicado para hemofilia B, que chega a custar U$ 3,5 milhões (R$ 17,4 milhões). Hoje, custa em torno de R$ 6 milhões – o preço, explica a fabricante Novartis, é definido pela Câmara de Regulação do Mercado de Medicamentos (CMED).
Após a aprovação dos pedidos das indústrias farmacêuticas fabricantes para registro dos novos medicamentos no Brasil pela Agência Nacional de Vigilância Sanitária (Anvisa), a Comissão Nacional de Incorporação de Tecnologias no Sistema Único de Saúde (Conitec) já deu parecer favorável para que Risdiplam e Zolgensma sejam ofertados na rede pública de saúde.
No entanto, os medicamentos ainda não foram incorporados pelo guia PCDT (Protocolos Clínicos e Diretrizes Terapêuticas), uma etapa exigida para que possam ser disponibilizados aos pacientes pelo SUS. Com isso, é necessário que as famílias recorreram à judicialização para obrigar o SUS a fornecer as medicações.
Zolgensma, por exemplo, foi incorporado pela Conitec para o tratamento de crianças com AME do tipo 1 com até seis meses de idade no final de 2022. Essa terapia gênica promissora deveria estar disponível no SUS em junho de 2023, mas ainda não é oferecida. No entanto, a comercialização depende da atualização do Protocolo Clínicos e Diretrizes Terapêuticas (PCDT), que está em fase de elaboração no Ministério da Saúde.
O PCDT atual ainda não contempla a inclusão de Zolgensma, porém, uma nova atualização do PCDT está em elaboração pelo Ministério da Saúde e aguardamos a publicação deste documento”, informou a indústria farmacêutica Novartis ao Portal ViDA & Ação.
Ainda segundo a fabricante “conforme previsto em lei, o prazo dos 180 dias previstos para a disponibilização do medicamento no SUS, após parecer positivo da Conitec encerrou-se no início de junho de 2023″.
AME: uma doença progressiva e sem cura
A atrofia muscular espinhal (AME) é uma doença rara grave que não tem cura. A incidência é de aproximadamente 1 a cada 10.000 nascidos vivos. No Brasil, o número se aproxima de 50 a 60 novas crianças a cada ano, segundo a neurofisiologista e neurologista Marcela Câmara Machado. Na Bahia, são entre 7 e 8 crianças nascidas com a doença, por ano. “É um número alto para uma doença tão complexa”.
Já de acordo com a Associação Brasileira de Amiotrofia Espinhal (Abrame), o país tem hoje cerca de 300 novos casos de AME por ano. Segundo a Anvisa, entre 45% e 60% das crianças acometidas com a AME desenvolvem a forma mais grave (tipo 1). A doença pode evoluir para a morte, sendo a principal causa de falecimentos em crianças por causa de uma enfermidade monogenética.
A doença é causada pela pela alteração (deleção/mutação) do gene SMN1, uma proteína necessária para os neurônios ligados ao movimento dos músculos. Sem um gene SMN1 funcional, crianças com AME tipo 1 rapidamente perdem os neurônios motores responsáveis pelas funções musculares. A AME produz atrofia progressiva dos músculos, dificultando a condição de movimentação dos pacientes.
“Os indivíduos nascem com uma alteração genética que perde os neurônios, nervos responsáveis pelos movimentos, que estão na região da medula”. A alteração genética faz com que não haja produção de uma substância que deixa esse neurônio saudável.
De acordo com o Ministério da Saúde, “é uma doença degenerativa, passada de pais para filhos e que interfere na capacidade do corpo de produzir uma proteína essencial para a sobrevivência dos neurônios motores, responsáveis pelos gestos voluntários vitais simples do corpo, como respirar, engolir e se mover (andar)”.
Sintomas de AME em cada estágio da doença
Marcela explicou que na fase um, a criança tem uma perda completa desses neurônios, tem uma fraqueza progressiva que leva a uma insuficiência respiratória. “E essa criança, se não tratada, falece até um ano de vida. Ela precisa de ventilação. Se a gente der suporte ventilatório, ela fica dependente de ventilação da parte respiratória durante toda a vida.
No estágio do tipo dois, a criança começa a ter sintomas entre seis meses e 18 meses. O tipo três é acima de 18 meses”. A criança começa uma fraqueza progressiva a partir dessa idade. O desfecho não é mais ventilatório.
No tipo 2, depois de 6 meses, a criança não é capaz de andar; fica o tempo todo na cadeira de rodas. Na fase da adolescência, ela tem necessidade de suporte ventilatório. É uma doença muito grave, com impactos social, emocional, psíquico muito grandes nas famílias e no paciente também”, explica a neurofisiologista e neurologista.
Sobre o Universo Coletivo AME
O Universo Coletivo AME é a maior coalizão no Brasil pela causa da atrofia muscular espinhal. O grupo atua no acolhimento, educação, conscientização e em ações voltadas para políticas públicas e visa acelerar a cobertura da AME no Teste do Pezinho, para que o diagnóstico seja precoce e, também, para garantir o acesso de todos os pacientes aos medicamentos disponíveis no SUS.
O Coletivo foi fundado em 2019 pela união de cinco instituições que atuam há mais de 20 anos em diferentes regiões do país e são lideradas por mães que vivenciam a AME no dia a dia. São elas: Associação de Doenças Neuromusculares Donem), Instituto Viva Íris, Instituto Nacional da Atrofia Muscular Espinhal (Iname), Instituto Fernando, e Associação Brasileira de Amiotrofia Espinhal (Abrame).
Em julho de 2023, o Senado Federal instituiu o 8 de agosto como Dia Nacional da Pessoa com Atrofia Muscular Espinhal.



